SpaMTP: Single Cell Multi-Omic Analysis (Metabolomics, Transcriptomics, Proteomics)
Source:vignettes/human_brain_tissue_analysis.Rmd
human_brain_tissue_analysis.RmdProteomics + Spatial Metabolomics Integration
This tutorial demonstrates how to explore protein intensities alongside spatial metabolomics and transcriptomics within a single SpaMTP object. The dataset is derived from a patient with low-grade glioma (LGG), with tissue curated to retain a clear distinction between the leading edge and tumour regions. The workflow covers QC maps, metabolite annotation, multi-panel spatial plots, and pathway scores that combine metabolite and RNA signals.
Requirements
You will need a SpaMTP object that contains:
-
SPMassay with m/z features. -
SPTassay with gene counts. - Metadata columns for spatial coordinates (
x_centroid,y_centroid). - Optional protein intensity columns (e.g.,
GFAP_intensity).
The dataset used in this tutorial consists of aligning SM, ST and SP data on serial section. This was achieved using the python package SMINT. The alignment script and python-to-SpaMTP script can be found following the respective links.
Load the integrated object
data <- readRDS(url("https://zenodo.org/records/18282678/files/brain_tumour_integrated.rds?download=1"))
cregpathway <- readRDS(url("https://zenodo.org/records/18282678/files/brain_tumour_combined_pathways.rds?download=1"))Below we can see the structure of our integrated data object
data## An object of class Seurat
## 913 features across 37886 samples within 2 assays
## Active assay: SPM (574 features, 574 variable features)
## 3 layers present: counts, data, scale.data
## 1 other assay present: SPT
## 7 dimensional reductions calculated: PCA, UMAP, spt.pca, spt.umap, spm.pca, spm.umap, wnn.umap
## 2 images present: slice1, image
SpatialFeaturePlot(
data,
features = c("nCount_originalexp", "nFeature_originalexp"),
pt.size.factor = 2
) & theme(legend.position = "right",legend.direction = "vertical")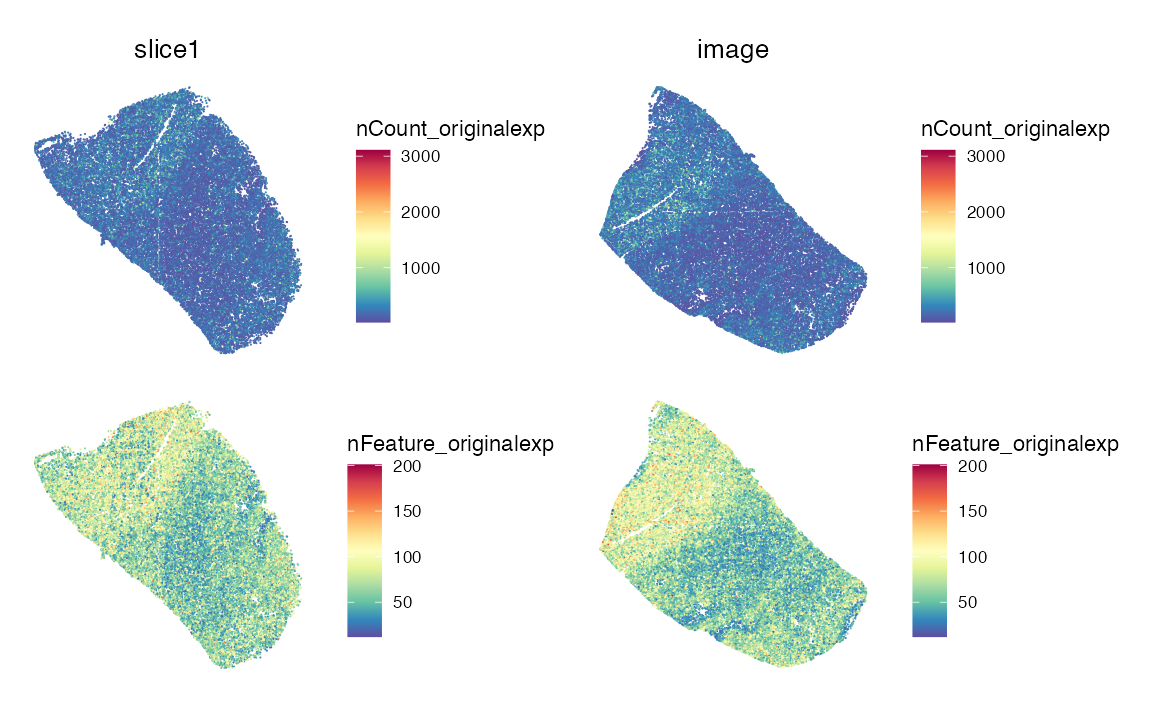
Annotate m/z features (optional)
We extract m/z values from the SPM assay and run a basic
annotation lookup.
spm <- data[["SPM"]]
mz_vals <- as.numeric(sub("^mz-", "", rownames(spm)))
spm@meta.features$raw_mz <- mz_vals
data[["SPM"]] <- spm
mz_df <- data.frame(
row_id = seq_along(mz_vals),
mz = mz_vals
)
annoresult <- SpaMTP:::annotateTable(mz_df, db = rbind(HMDB_db, Chebi_db))
head(annoresult)| ID | Match | observed_mz | Reference_mz | Error | Adduct | Formula | Exactmass | Isomers | InchiKeys | IsomerNames | Isomers_IDs | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M+H.1 | 1 | TRUE | 71.07229 | 71.07241 | 1.653058 | M+H | C4H8N | 70.06513 | CHEBI:36781 | ZVJHJDDKYZXRJI-UHFFFAOYSA-O | 1-pyrrolinium | chebi:36781 |
| M+H.2 | 3 | TRUE | 72.08015 | 72.08078 | 8.613587 | M+H | C4H9N | 71.07350 | HMDB0031641 | RWRDLPDLKQPQOW-UHFFFAOYSA-N | Pyrrolidine | hmdb:HMDB0031641 |
| M+H.3 | 3 | TRUE | 72.08015 | 72.08078 | 8.613601 | M+H | C4H9N | 71.07350 | HMDB0243874 | UJGVUACWGCQEAO-UHFFFAOYSA-N | 1-Ethylaziridine | hmdb:HMDB0243874 |
| M+H.4 | 3 | TRUE | 72.08015 | 72.08078 | 8.623396 | M+H | C4H9N | 71.07350 | CHEBI:31108; CHEBI:33135 | ASVKKRLMJCWVQF-UHFFFAOYSA-N; RWRDLPDLKQPQOW-UHFFFAOYSA-N | 3-buten-1-amine; pyrrolidine | chebi:31108; chebi:33135 |
| M+H.5 | 4 | TRUE | 74.09586 | 74.09643 | 7.687876 | M+H | C4H11N | 73.08915 | HMDB0031321; HMDB0032179; HMDB0034198; HMDB0041878 | HQABUPZFAYXKJW-UHFFFAOYSA-N; BHRZNVHARXXAHW-UHFFFAOYSA-N; KDSNLYIMUZNERS-UHFFFAOYSA-N; HPNMFZURTQLUMO-UHFFFAOYSA-N | 1-Butylamine; sec-Butylamine; 2-Methyl-1-propylamine; Diethylamine | hmdb:HMDB0031321; hmdb:HMDB0032179; hmdb:HMDB0034198; hmdb:HMDB0041878 |
| M+H.6 | 4 | TRUE | 74.09586 | 74.09643 | 7.687889 | M+H | C4H11N | 73.08915 | HMDB0255263 | DAZXVJBJRMWXJP-UHFFFAOYSA-N | N,N-DIMETHYLETHYLAMINE | hmdb:HMDB0255263 |
Multi-panel spatial maps (cell type, gene, protein, metabolite)
Pick a metabolite m/z, a gene, and a protein feature available in
your object. We will use the protein GFAP as an example. The
corresponding gene and mz values are AQP4 and
mz-146.1651 respectfully.
p1 <- SpatialDimPlot(data, group.by = "Anno4.1", images = "image", pt.size.factor = 2.5) +scale_fill_viridis_d(option = "turbo")+guides(fill = guide_legend(ncol = 2))
p2 <- SpatialFeaturePlot(data, features = "AQP4", slot = "data",images = "image",pt.size.factor = 2.5)+ scale_fill_viridis_c(option = "magma", na.value = "lightgrey")
p3 <- SpatialFeaturePlot(data, features = "GFAP_intensity", images = "image",pt.size.factor = 2.5)+ scale_fill_viridis_c(option = "magma")
p4 <- SpatialMZPlot(data, mzs = 146.1651, images = "image",pt.size.factor = 2.5, assay = "SPM", slot = "data")+ scale_fill_viridis_c(option = "magma")
(p1|p2)/(p3|p4) & coord_flip() & scale_y_reverse() & theme(legend.position = "right",legend.direction = "vertical")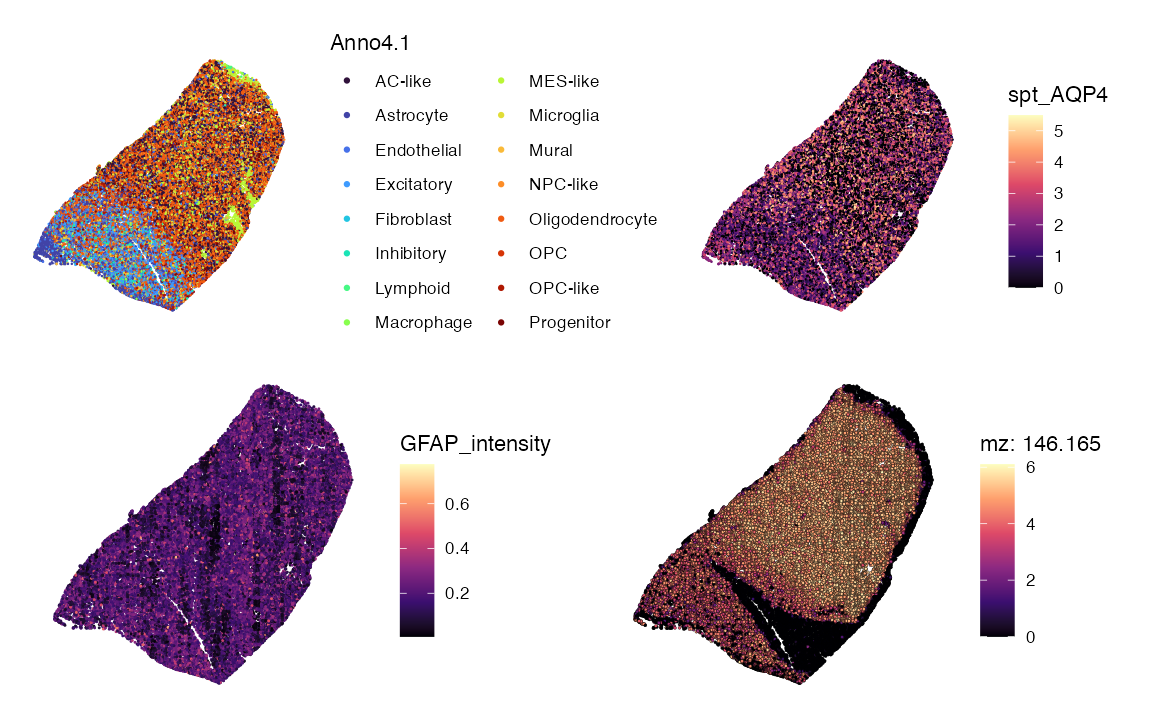
Pathway-level metabolite, RNA, and combined scores
We compute pathway scores for selected pathways by binning m/z features and summing leading-edge genes, then combine the two modalities.
min_max <- function(x) {
rng <- range(x, na.rm = TRUE)
if (is.infinite(rng[1]) || rng[1] == rng[2]) {
return(rep(0, length(x)))
}
(x - rng[1]) / (rng[2] - rng[1])
}
sum_genes <- function(obj, genes) {
counts <- obj[["SPT"]]@counts
valid <- intersect(genes, rownames(counts))
if (length(valid) == 0) {
warning("No genes found for pathway in SPT counts")
return(rep(0, ncol(counts)))
}
colSums(counts[valid, , drop = FALSE])
}
score_pathway <- function(obj, pathway_df, pathway_name, type = "wiki", weight = 0.6) {
selected <- pathway_df[
pathway_df$pathwayName == pathway_name & pathway_df$type == type,
]
stopifnot(nrow(selected) > 0)
mzs <- as.numeric(strsplit(gsub("\\[.*?\\]", "", selected$adduct_info[1]), ";")[[1]])
mzs <- unlist(lapply(mzs, function(x) FindNearestMZ(obj, target_mz = x, assay = "SPM")))
obj <- BinMetabolites(
obj,
mzs,
slot = "counts",
bin_name = paste0(pathway_name, "_met"),
assay = "SPM"
)
genes <- strsplit(selected$leadingEdge_genes[1], ";")[[1]]
obj@meta.data[[paste0(pathway_name, "_rna")]] <- sum_genes(obj, genes)
met_norm <- min_max(obj@meta.data[[paste0(pathway_name, "_met")]])
rna_norm <- min_max(obj@meta.data[[paste0(pathway_name, "_rna")]])
obj@meta.data[[paste0(pathway_name, "_combined")]] <-
weight * met_norm + (1 - weight) * rna_norm
list(obj = obj, genes = genes)
}
pathway_name <- "Glucose metabolism"
pathway_type <- "wiki"
res <- score_pathway(data, cregpathway, pathway_name, type = pathway_type)
data <- res$obj
p1 <- SpatialFeaturePlot(data, features = paste0(pathway_name, "_met"), pt.size.factor = 2.5)
p2 <- SpatialFeaturePlot(data, features = paste0(pathway_name, "_rna"), pt.size.factor = 2.5)
p3 <- SpatialFeaturePlot(data, features = paste0(pathway_name, "_combined"), pt.size.factor = 2.5)
p1/p2/p3 & theme(legend.position = "right",legend.direction = "vertical")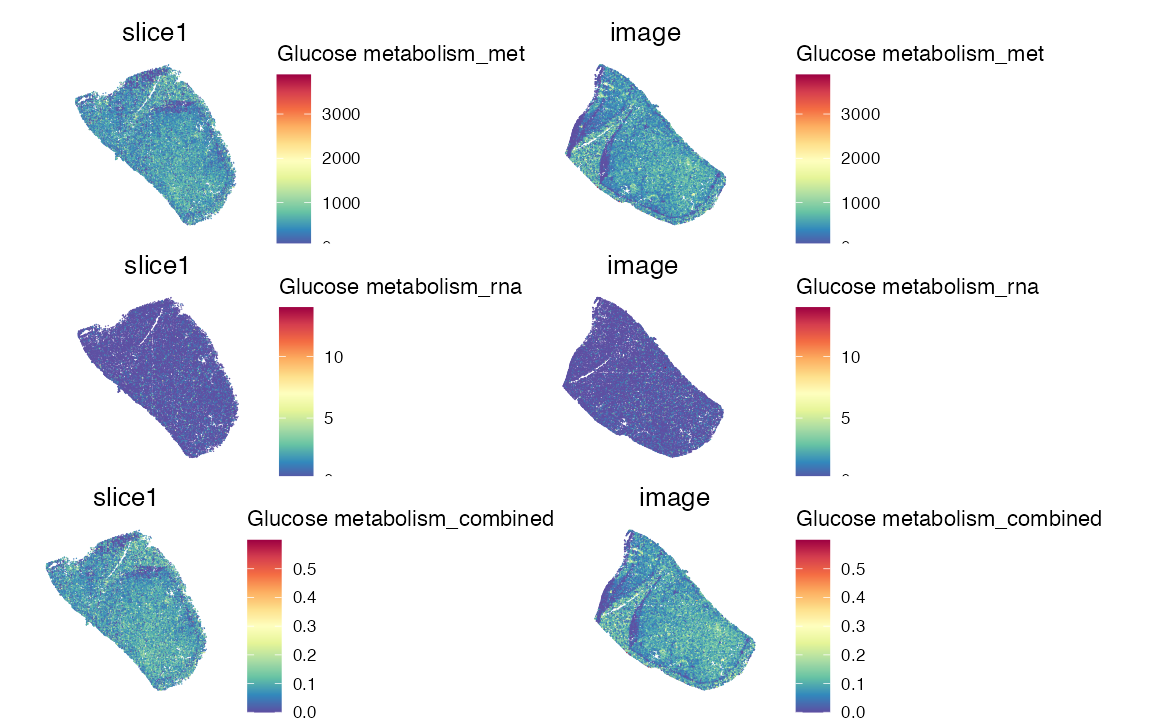
pathway_name <- "Neuronal System"
pathway_type <- "Reactome"
res <- score_pathway(data, cregpathway, pathway_name, type = pathway_type)
data <- res$obj
p1 <- SpatialFeaturePlot(data, features = paste0(pathway_name, "_met"), pt.size.factor = 2.5)
p2 <- SpatialFeaturePlot(data, features = paste0(pathway_name, "_rna"), pt.size.factor = 2.5)
p3 <- SpatialFeaturePlot(data, features = paste0(pathway_name, "_combined"), pt.size.factor = 2.5)
p1/p2/p3 & theme(legend.position = "right",legend.direction = "vertical")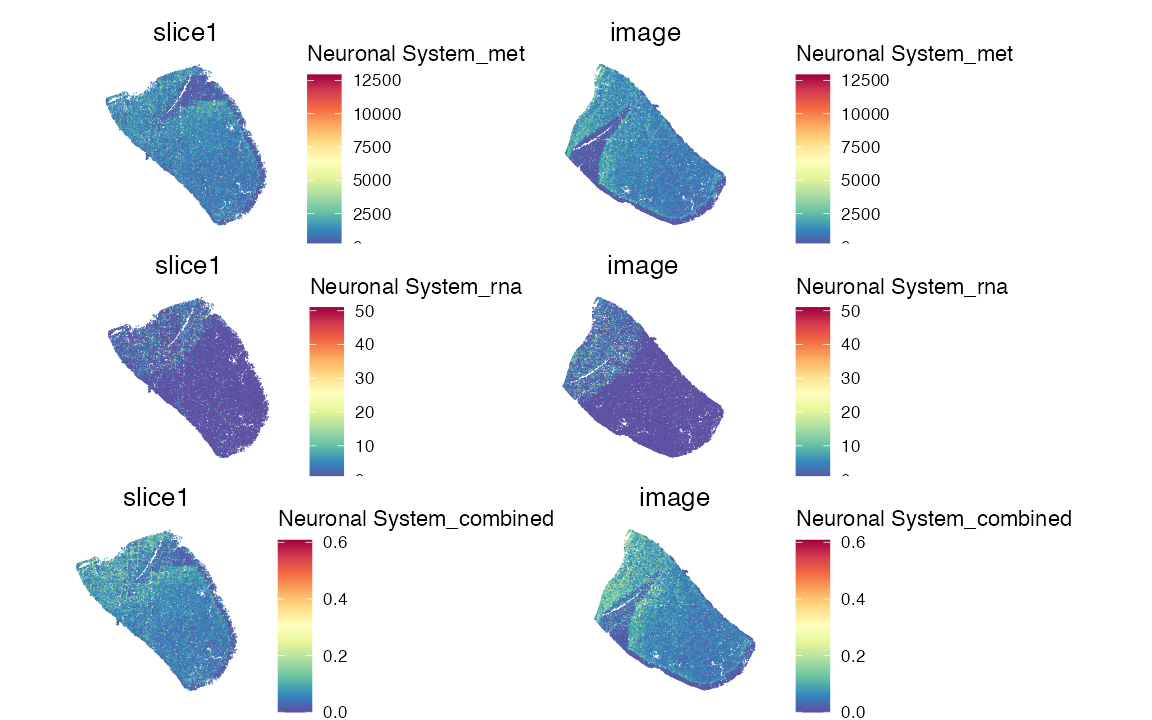
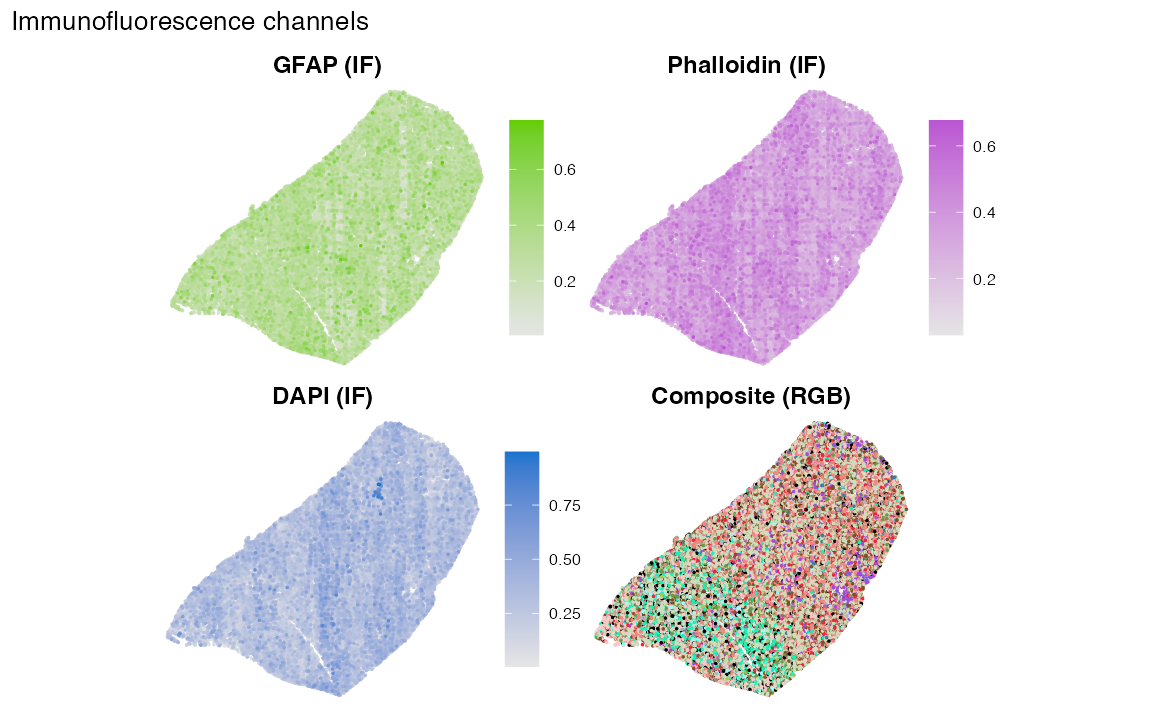
Optional: plot immunofluorescence channels
If your metadata includes separate intensity channels and an RGB composite, you can map them as additional spatial panels.
x_coord <- data@meta.data[["x_centroid"]]
y_coord <- data@meta.data[["y_centroid"]]
required_cols <- c("GFAP_intensity", "Phalloidin_intensity", "DAPI_intensity", "color")
if (all(required_cols %in% colnames(data@meta.data))) {
parse_rgb <- function(txt) {
vapply(txt, function(s) {
if (is.na(s) || s == "") return(NA_character_)
nums <- as.numeric(strsplit(gsub("[() ]", "", s), ",", fixed = TRUE)[[1]])
if (length(nums) != 3 || any(is.na(nums))) return(NA_character_)
rgb(nums[1], nums[2], nums[3])
}, character(1))
}
rgb_hex <- parse_rgb(data@meta.data[["color"]])
chan_df <- data.frame(
x = x_coord,
y = y_coord,
gfap_if = data@meta.data[["GFAP_intensity"]],
phall_if = data@meta.data[["Phalloidin_intensity"]],
dapi_if = data@meta.data[["DAPI_intensity"]],
rgb_hex = rgb_hex
)
chan_panel <- function(var, title, high_col) {
df_ord <- chan_df[order(chan_df[[var]]), ]
ggplot(df_ord, aes(x, y, colour = .data[[var]])) +
geom_point(size = 0.25) +
coord_equal() +
scale_colour_gradient(
low = "grey90",
high = high_col,
na.value = "grey90",
guide = "colourbar"
) +
guides(colour = guide_colourbar(barheight = unit(38, "mm"))) +
labs(title = title, colour = NULL) +
theme_void(base_size = 10) +
theme(plot.title = element_text(hjust = 0.5, face = "bold"))
}
p_gfap <- chan_panel("gfap_if", "GFAP (IF)", high_col = "chartreuse3")
p_phall <- chan_panel("phall_if", "Phalloidin (IF)", high_col = "mediumorchid")
p_dapi <- chan_panel("dapi_if", "DAPI (IF)", high_col = "dodgerblue3")
p_rgb <- ggplot(chan_df, aes(x, y, colour = rgb_hex)) +
geom_point(size = 0.25) +
coord_equal() +
scale_colour_identity() +
labs(title = "Composite (RGB)") +
theme_void(base_size = 10) +
theme(plot.title = element_text(hjust = 0.5, face = "bold"))
(p_gfap | p_phall) / (p_dapi | p_rgb) +
plot_annotation(title = "Immunofluorescence channels") &
theme(legend.box = "vertical")
} else {
warning("Missing IF columns: ", paste(setdiff(required_cols, colnames(data@meta.data)), collapse = ", "))
}